Background
Progressive muscular dystrophies are clinically, genetically, and biochemically heterogeneous group of non-inflammatory diseases, which are based on a primary muscle fiber defect accompanied by characteristic, but often non-pathognomonic pathological signs [1]. Clinically, they are characterized by progressive muscle weakness that affects the muscles of the limbs, axial muscles, and facial muscles to varying degrees.
In the past, muscular dystrophies have been classified according to the underlying clinical findings and the age of onset. Most of the clinical forms of muscular dystrophies manifest in the first decade of life, they are progressive and incurable [2]. An improved understanding of the mechanisms that underlie these forms has provided new clues about their classification [3–5]. Based on the world developments in the field of the molecular diagnostics, the clinical and molecular genetic classification of muscular dystrophies was first published in the Neuromuscular Disorders journal (1999, 9th issue). Currently, progressive muscular dystrophies are classified on a genetic basis [6, 7].
As a separate disorder, muscular dystrophies are relatively rare. However, they represent a significant proportion of neuromuscular diseases, with a general prevalence of 16.14 per 100,000 [8]. There are differences in the prevalence of muscular dystrophy due to genetic differences between populations and ethnic groups, as well as differences in the availability of molecular diagnostics. Also, the availability of medical information sources differs depending on the country and region. In addition, some patients may not seek medical attention at all and therefore undiagnosed cases lead to an underestimation of the true prevalence of muscular dystrophy.
No one disputes the important role of obtaining a medical history and clinical investigation in patients with neuromuscular diseases, and in particular in those with muscular dystrophy. The combination of clinical signs and analysis of the possible type of inheritance allows one to suspect specific forms of muscular dystrophy and determine the direction of further research. Although the coincidences between genetically distinguishable forms complicate the diagnostic search. Clinicians increasingly need to rely on electrophysiological, imaging, and genetic data for more accurate differential diagnosis.
Electromyography (EMG) is a significant method for early diagnosis of muscular dystrophies, including diffe-rential diagnosis of neuromuscular pathology, determining the stage and activity of the myodystrophic process. However, EMG changes do not have high specificity [9, 10]. For progressive muscular dystrophies, a large number of low-amplitude fibrillation potentials are characteristic, which arise as a result of reinnervation of functionally incapacitated muscle fibers. Positive sharp waves appear as a result of the death of muscle fibers. In hypertrophied muscles, pseudomyotonic discharges appear in the form of groups of fibrillations [11]. Dystrophic muscle lesions are more likely to induce myopathic activity on EMG than non-dystrophic lesions [12].
There is a growing number of evidence proving that muscle imaging can play an important role in identifying genetically distinct conditions, assisting clinicians in selecting appropriate genetic diagnostic tests as well as the muscles to target for pathological examinations. Although, muscle imaging allows for the identification of specific patterns of muscle damage and can be used in the differential diagnosis of muscular dystrophy forms [13–16]. Muscle magnetic resonance imaging (MRI) has a higher diagnostic value in the early diagnosis of slowly progressive diseases in which selective patterns of muscle involvement can be detected over a long period [17]. Muscle MRI can be used as a diagnostic tool to describe the degree and nature of muscle damage and to determine the localization of future muscle biopsy, if necessary [18–20]. This can help in narrowing the diagnostic search and differential diagnosis of muscular dystrophy forms.
At the moment, there is no etiopathogenetic treatment for progressive forms of muscular dystrophies, except for certain forms, such as Duchenne muscular dystrophy. Currently, medical treatment is prescribed taking into account the results of clinical and instrumental examinations and concomitant pathology (cardio-, pneumopathy). Also, the management of patients is complex and includes pathogenetic and special therapy, taking into account the severity of the disease (mild, moderate, severe), stages (compensation, subcompensation, decompensation), physiotherapy procedures, singlet oxygen therapy, physiotherapy exercises (breathing exercises, stretch gymnastics), special diet, electroacupuncture, gentle massage of functionally intact muscles [21]. Verification of individual forms of muscular dystrophies is important not only for confirming the diagnosis in patients and genetic prevention in families (diagnosis of heterozygous carriage), but also for preclinical diagnosis for early observation and, if possible, treatment. However, molecular genetic diagnostics is not always available to patients, which delays the time of diagnosis.
The purpose of the research: to identify MRI signs of muscle damage in muscular dystrophies, to compare the revealed changes with EMG data, laboratory, and genetic examination results.
Materials and methods
We performed MRI of the muscles of the lower extremities in 17 patients with muscular dystrophy. Seven healthy individuals formed a control group. The examination was carried out on a Signa HDxt 1.5T MRI scanner (General Electric, USA) with a magnetic field strength of 1.5 Tesla, using a soft surface coil. The protocol included T1, T2 and STIR modes in axial and coronary projections, using anatomical landmarks. The examination was carried out at the level of the hips on both sides, without contrast enhancement. The study time was 20–30 minutes for each patient. Evaluation of the obtained images was carried out by visual assessment of the signal from the muscles of the lower extremities for each patient according to the 5-point Mercuri scale (2002). On the scans of the thighs, we have examined the muscles of the anterior (m.rectus femoris, m.sartorius, m.vastus lateralis, m.vastus intermedius, m.vastus medialis), posterior (m.semimembranosus, m.semitendinosus, m.biceps femoris caput longum, m.biceps femoris caput breve) and medial compartments (m.adductor magnus, m.brevis, m.longus, m.gracilis, m.pectineus).
All patients underwent a preliminary EMG (needle) and the level of creatine kinase was assessed, followed by a genetic analysis using the next-generation sequencing (NGS). Also, each patient was examined for impaired ability to self-care using the Barthel scale and the grade was determined by the 10-point Vignos scale, corresponding to the patient’s motor activity.
Results and discussion
Our study included male (n = 7) and female (n = 10) participants aged 20 to 46 years. All patients applied to the Regional Center of Neurosurgery and Neurology in Uzhhorod from 2016 to 2020. The average age at onset and diagnosis was 15.3 and 29.8 years, respectively. The median number of years took to make a diagnosis was 15. The most common initial symptoms were gait disturbance, difficulty running, climbing stairs and raising arms, weakness of the lower extremities. Other symptoms included facial muscle weakness (1 patient — 5.9 %) and general hypotension. In 3 people (17.6 %), a contracture of the Achilles tendon was revealed. Two individuals (11.8 %) required a wheelchair, two patients (11.8 %) could walk with assistance, 13 people (76.4 %) could walk independently, without assistance. On the 10-point Vignos scale, one patient had grade 1 (5.9 %), another one had grade 2 (5.9 %), 9 — grade 3 (52.9 %), 2 — grade 4 (11.8 %), 3 — grade 5 (17.6 %) and one patient — grade 8 (5.9 %).
A direct physical examination of the muscles was also performed and included Gower sign, 4-stair climb, and Barthel index. 11.8 % of study participants were not able to get up from the floor; 17.6 % were not able to get up without assistance; 53 % had the classic Gower sign; and only 5.9 % climbed without problems. Climbing four stairs was difficult for almost all participants (14 of 17), two patients couldn’t do it at all, and only one had no problems with it. According to the Barthel index, 3 patients (17.6 %) had a significant dependence (up to 60 points) and 14 (82.4 %) — a moderate dependence (up to 91 points).
There is a relationship between the duration of the di-sease and the degree of self-care loss. The shorter the period of illness in years was observed in patients, the less was the degree of impairment of the ability to self-care according to the Barthel scale (Fig. 1).
Serum creatine kinase levels were measured in all patients. There was a moderate increase (up to 5-fold) in 7 patients (41.2 %), in 3 (17.6 %) this indicator was within the normal range, and 7 people (41.2 %) reported a significant increase (10-fold or higher). Electromyography revealed changes mainly by myopathic type in 16 cases (94.1 %), one patient (5.9 %) had a neuronal type, and 2 (11.8 %) of 17 people also had fibrillations and fasciculations. Of 17 patients, 5 (29.4 %) had cardiomyopathy and 1 (5.9 %) — symptomatic arrhythmia.
Magnetic resonance imaging showed damage to the muscles of the thigh from grade 1 to grade 4 according to Mercuri in 16 patients (94.1 %), while one person had not any changes. So, muscle MRI was performed, which showed lesions of mm.peroneus longus, gastrocnemius caput mediale. The lesions of the muscles in the posterior compartment of the thigh were as follows: in 13 of 16 patients, m.semimembranosus and m.semitendinosus were affected, in 15 — m.biceps femoris caput longum, and in 14 — m.biceps femoris caput breve. The imaging of the muscles of the medial compartment showed that the pectineus muscle remained preserved in 14 individuals, 10 people had lesions of m.gracilis and m.adductor brevis, 12 — of m.adductor longus and 9 patients — of m.adductor magnus. The lesions of the muscles of the anterior compartment were as follows: m.rectus femoris was affected in 5 patients, m.sartorius — in 12, m.vastus lateralis — in 15, m.vastus intermedius — in 14, and m.vastus medialis — in 13 people.
A different degree of muscle damage according to the Mercuri classification is presented in Fig. 2.
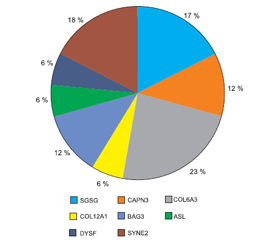
The genetic testing by the NGS method showed that three people had mutations in the SGSG gene, two — in the CAPN3, four — in COL6A3, one — in COL12A1, two — in BAG3, one patient each — in ASL and DYSF genes, and three more patients — in SYNE2 (Fig. 3).
Evaluation of patterns of muscle involvement in diffe-rent types of muscular dystrophy provided important observations (Table 1). Several muscles of the thigh (the long and short heads of the biceps femoris, the vastus lateralis muscle, the semimembranosus and semitendinosus muscles, and the adductor longus muscle) were most frequently involved in all studies. In almost all patients, the preservation of the pecti-neus and gracilis muscles was most often noted. It was found that the m.sartorius and m.rectus femoris, m.vastus lateralis and m.vastus medialis, m.adductor magnus and m.adductor brevis are characterized by a combination of damage (from involved to intact) in various diseases.
Muscle involvement was specific for a particular gene mutation (Fig. 4).
Muscle MRI in mutations in various genes showed the greatest involvement of the following thigh muscles:
— in the COL6A3 gene — of the m.vastus med., m.vastus lat., m.vastus int., m.gracilis, m.adductor long., m.semimembranosus, m.semitendinosus, m.biceps femoris (Fig. 4A);
— in the CAPN3 gene — of m.vastus med., m.vastus lat., m.vastus int., m.semimembranosus, m.semitendinosus, m.biceps femoris, caput long., m.adductor long., m.adductor brevis, and m.adductor magnus (Fig. 4B);
— in the SGSG gene — of m.adductor long., m.semimembranosus, m.semitendinosus, m.biceps femoris (Fig. 4C);
— in the SYNE2 gene — of m.sartorius, m.vastus med., m.vastus int., m.gracilis, m.biceps femoris (Fig. 4D);
— in the BAG3 gene — of m.adductor long., m.adductor brevis and m.adductor magnus, m.semimembranosus, m.vastus med., m.vastus int., m.biceps femoris (Fig. 4E);
— in the ASL gene — of m.rectus femoris, m.vastus lat., m.adductor long., m.adductor brevis, m.adductor magnus, m.semimembranosus, m.semitendinosus, m.biceps femoris (Fig. 4F).
Visual examination of the tomograms of the muscles of the lower extremities in the control group did not reveal pathological changes in the T1, T2 and STIR modes: there was a normal intensity of the muscle signal (the signal was homogeneous, hypointense, well-contrasting with the subcutaneous and intermuscular fat), the muscles were clearly identified, the content of adipose tissue corresponded to normal values.
Conclusions
We have noted the role of magnetic resonance imaging as a highly informative method for diagnosing muscle pathology. MRI can detect early involvement of the thigh muscles. We showed that 94.1 % of patients had a lesion of the muscles of the posterior thigh, 64.7 % — in the medial compartment, and in 70.6 % of cases, there was a lesion of the muscles of the anterior compartment of the thigh. We also showed the difference between the lesions of certain muscle groups in different types of muscular dystrophy, which significantly narrows the differential diagnostic search, simplifying genetic testing. All patients had an increased level of creatine kinase in the blood serum, and 94.1 % had changes by myopathic type when performing EMG. Also, there was a relationship between the duration of the disease and the degree of loss of self-care.
Thus, accurate nosological diagnosis of muscular dystrophies is possible only with the use of modern diagnostic methods, such as laboratory, electrophysiological, radiological and genetic ones. However, given the socio-economic situation of the majority of patients, limited access to genetic testing, it is important to develop screening methods for the diagnosis of early manifestations of progressive muscular dystrophy, including the identification of specific patterns of muscle damage. In turn, accurate diagnosis of muscular dystrophy forms is the ground for the next most important step — the development of effective treatment methods.
Received 18.03.2021
Revised 03.04.2021
Accepted 12.04.2021
Список литературы
1. Emery A. The muscular dystrophies. Lancet. 2002. 359. 687-95. doi: 10.1016/S0140-6736(02)07815-7.
2. Евтушенко С., Шаймурзин М., Евтушенко О., Евтушенко И. Нервно-мышечные заболевания у детей: Монография. Донецк: Изд-во «Ноулидж» (донецкое отделение), 2014. 218 с.
3. Mercuri E., Muntoni F. The ever-expanding spectrum of congenital muscular dystrophies. Ann. Neurol. 2012. 72. 9-17. doi: 10.1002/ana.23548.
4. Guglieri M., Straub V., Bushby K., Lochmuller H. Limb-girdle muscular dystrophies. Curr. Opin. Neurol. 2008. 21(5). 576-84. doi: 10.1097/WCO.0b013e32830efdc2.
5. Muntoni F., Voit T. The congenital muscular dystrophies in 2004: a century of exciting progress. Neuromuscul. Disord. 2004. 14(10). 635-49. doi: 10.1016/j.nmd.2004.06.009.
6. Wicklund M. The muscular dystrophies. Continuum (Minneap. Minn.). 2013. 19(6 Muscle Disease). 1535-1570. doi: 10.1212/01.con.0000440659.41675.8b.
7. Narayanaswami P., Weiss M., Selcen D. et al. Evidence-based guideline summary: Diagnosis and treatment of limb-girdle and distal dystrophies. Neurology. 2014. 83(16). 1453-63. doi: 10.1212/WNL.0000000000000892.
8. Mah J., Korngut L., Fiest K.M. et al. A systematic review and meta-analysis on the epidemiology of the muscular dystrophies. Can. J. Neurol. Sci. 2016. 43(1). 163-77. doi: 10.1017/cjn.2015.311.
9. Miller T. Differential diagnosis of myotonic disorders. Muscle and Nerve. 2008. 37(3). 293-299. doi: 10.1002/mus.20923.
10. Hehir M., Logigian E. Electrodiagnosis of myotonic disorders. Phys. Med. Rehabil. Clin. N. Am. 2013. 24(1). 209-20. doi: 10.1016/j.pmr.2012.08.015.
11. Николаев С. Практикум по клинической электромиографии. Иваново: Ивановская государственная медицинская академия, 2003. 127-128.
12. Harper C. Congenital myopathies and muscular dystrophies. In: Brown W.F., Bolton C.F., Aminoff M.J. (eds). Neuromuscular function and disease. Philadelphia: WB Saunders, 2002. 1355-1374. doi: 10.1055/s-2008-1062269.
13. Mercuri E., Pichiecchio A., Allsop J., Messina S., Pane M., Muntoni F. Muscle MRI in inherited neuromuscular disorders: past, present, and future. J. Magn. Reson. Imaging. 2007. 25(2). 433-40. doi: 10.1002/jmri.20804.
14. Палагута Г., Смоланка В., Орос М., Дутка І. Діагностична роль магнітно-резонансної томографії м’язів при нервово-м’язових захворюваннях (науковий огляд та особисте спостереження). Международный неврологический журнал. 2019. 1(103). 26-31. doi: 10.22141/2224-0713.1.103.2019.158635.
15. Palahuta H., Fartushna O. The role of magnetic resonance imaging of muscles in the differential diagnosis of certain forms and subtypes of limb-girdle muscular dystrophy: case analysis. International Neurological Journal. 2020. 16(8). 43-47. doi: 10.22141/2224-0713.16.8.2020.221960.
16. Палагута Г., Смоланка В., Дутка І. Діагностика поясної складнокурабельної міопатії з використанням методу МРТ (науковий огляд і особисте спостереження). Международный неврологический журнал. 2020. 16(2). 13-17. doi: 10.22141/2224-0713.16.2.2020.200958.
17. Jungbluth H. Myopathology in times of modern imaging. Neuropathol. Appl. Neurobiol. 2017. 43(1). 24-43. doi: 10.1111/nan.12385.
18. Wattjes M., Kley R., Fischer D. Neuromuscular imaging in inherited muscle diseases. Eur. Radiol. 2010. 20(10). 2447-60. doi: 10.1007/s00330-010-1799-2.
19. Ten Dam L., van der Kooi A., Verhamme C., Wattjes M., de Visser M. Muscle imaging in inherited and acquired muscle diseases. Eur. J. Neurol. 2016. 23(4). 688-703. doi: 10.1111/ene.12984.
20. Quijano-Roy S., Carlier R., Fischer D. Muscle imaging in congenital myopathies. Semin. Pediatr. Neurol. 2011. 18. 221-229.
21. Евтушенко С., Шаймурзин М., Евтушенко О., Евтушенко Л., Дегонская Е., Евтушенко И., Сохань Д. Ранняя клинико-инструментальная диагностика и терапия быстро- и медленнопрогрессирующих мышечных дистрофий и амиотрофий. Международный неврологический журнал. 2007. 4(14). 47-63.

/53.jpg)
/54.jpg)
/53_2.jpg)
/54_2.jpg)